监测各类问题汇总解答

企业可以根据自身的设备情况以及消毒周期来确定具体的消毒和灭菌方式,并对其进行充分的验证。但一般情况下,不建议纯化水系统和注射用水系统仅仅采用紫外线定期消毒这一种方式。

洁净区嵌入式专用摄像头解决了该项难题。洁净区嵌入式专用摄像头安装于洁净区彩钢板墙体上,通过嵌入彩钢板的方式安装,安装高度距离天花板约 40-60cm。该摄像头平面为纯平面板,嵌入彩钢板后,摄像头平面与彩钢板平面平齐,在利用墙擦清洁墙面的同时实现了摄像头的清洁。同时,摄像头的纯平 面板零卫生死角,不存在清理不到位的情况。

关于洁净室技术标准,我国有多个国标均有所涉及,如:《医药工业洁净厂房设计规范》(GB50457-2008)、《洁净室施工及验收规范》(GB 50591-2010)等。悬浮粒子、浮游菌、沉降菌的检测方法可参考GB/T16292-2010、GB/T16293-2010、GB/T16294-2010等国标。
ISO14644对洁净室的技术标准也有很强参考意义,另外国家食品药品监管局药品认证管理中心组织编写的《药品GMP指南》中也列出了一些要求,如换气次数 D级动态标准:6-20次/h;C级动态标准:20-40次/h;B级动态标准:40-60次/h等。


清洁验证应当综合考虑设备使用情况、所使用的清洁剂和消毒剂、取样方法和位置以及相应的取样回收率、残留物的性质和限度、残留物检验方法的灵敏度等因素。
单一品种原料药生产过程中专用设备的清洁验证,从风险的角度看,专用设备上的活性成分残留对后续生产产品质量影响不大。清洁验证的重点在于确认是否存在相关杂质(降解产物、反应物)的残留,该残留是否能够确保药品的安全性和有效性。
如果为生产单一品种的专用设备,应综合评估原料药(或中间体)在相关设备上的性质,是否有高活性的杂质产生、该杂质在本清洁方式下残留的标准能否达到等。通常情况下,清洁验证需要通过取样检测的方式来证明,不能仅以目测无可见残留为指标。
目测残留无可见物料通常作为每次清洁行为之后的检测标准。
药品生产质量管理规范的要求是为了控制在取样过程中引入的污染和交叉污染风险。从风险角度看,取样的条件与被取样物料的生产条件一致不会放大污染和交叉污染的风险,所以取样区的空气洁净度级别应不低于所取样物料将被使用的生产条件是可以接受的。
虽然药品生产质量管理规范没有强制规定消毒剂必须轮换使用,但实际上对生产企业提出了更为科学的要求。企业以往通常是按照要求进行消毒剂轮换,而对消毒剂轮换使用对洁净区消毒的效果不做研究。新修订药品GMP要求企业对环境监测数据统计分析,进而确定不少于一类的消毒剂如何使用,最终确保消毒剂在洁净区内的消毒有效性。
企业采用何种防止污染和交叉污染的措施,应首先综合 考虑所生产药品的特性、工艺和预定用途等因素,进而确定厂房、生产设施和设备多产品共用的风险情形,再根据风险的级别进一步确定采取相应的措施将污染或交 叉污染降至可接受的水平,并应定期检查防止污染和交叉污染的措施并评估其适用性和有效性。
规范要求洁净厂房的压差梯度始终维持在10Pa以 上,主要是指在生产过程的控制,是为了防止在生产过程中由于空气流动产生的污染和交叉污染。在没有产品生产的过程中,企业可采取如设置值班风机或降低风机 频率等保持相对正压的措施,并进行风险评估,要有数据证明这种运行方式不会增加因洁净区压差改变而带来的微粒和微生物污染的风险。但一般而言,核心关键区 域与不同级别的压差不应小于10Pa。
企业在对系统进行设计、确认、运行、管理时需要考虑 多种因素对于生产环境的影响,并不能仅从一、两个方面考虑问题。如果企业的空气净化系统采取所问问题的方式,企业在进行验证时应考虑到停运的时间、环境的 温湿度、不同季节环境中可能存在的菌种、芽孢等最差条件,并进行充分验证。空气净化系统停运后重新开启,无额外的消毒措施,只单纯依靠自净时间控制,很多时候容易导致产品微生物污染的风险。
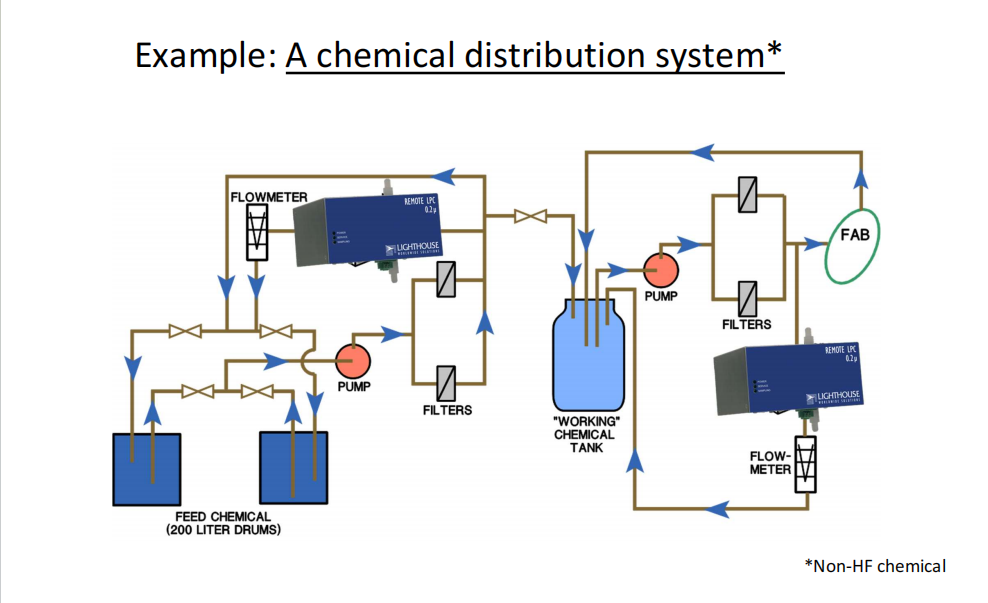
对原水水质定期监测的目的是为了保证制药工艺用水的质量。毋容置疑,企业使用的原水无论是地下水、地表水或市政供水,都应定期监测。如果是饮用水,其检测标准可以参照饮用水的国家标准;若原水并非饮用水,则需要企业自行制定相关标准(应当在厂房设施的DQ阶段建立而成,通过考察原水的质量进而指导纯化水制备的设计)。
同样原因,企业使用城市市政供水也应定期进行检测,从而及时发现原水水质的变化而采取应对措施,能够降低纯化水的质量风险。
由于环境监测所用培养基富含营养成分,容易长菌,如在生产区内配制、准备和培养,会极大增加药品生产过程中的微生物污染风险。因此,不建议在生产区内中间控制区进行洁净区环境监测的准备和培养。
选择甲醛熏蒸,还应当关注甲醛的残留,是否会对产品产生交叉污染,应当考虑使用后的恢复时间和残留量。企业应当根据自身实际情况综合评价后自行选择是否进行交替使用。
企业可以采取任何有效措施来使设备达到干燥状态,例如,使用压缩空气吹干等。在大多数情况下,应在清洗完毕尽快进行干燥,不能在潮湿状态下长时间存放,如果潮湿状态保持的时间过长,企业应当证明此保存时间的合理性。另外,企业在干燥措施的设计过程中应当考虑经济性和可操作性,并防止所用的干燥措施对已清洁的设备产生二次污染。
企业应基于日常维护、监测及验证和确认的情况来决定停产后HVAC系统的管理(包括测试的频率),并结合停产中的设施维护管理情况来确定是否需要重新验证。一般情况下,无菌产品生产环境应对浮游菌和沉降菌进行监测。对于口服固体制剂车间,可以根据生产质量管理的实际情况自行规定相关要求。但无论如何,企业均须确认HVAC系统停止使用一段时间重新启用后相关区域能够达到规定的洁净级别要求。


当生产质量稳定,不易被微生物污染且生产环节能够进行有效控制的产品时,考虑采用阶段性的生产方式组织生产。但必须经过全面的评估并制定规范的管理程序,且须完成相应的全部清洁验证工作。应明确规定出连续进行生产的批次和时间,换批次生产时,设备可以只进行中间清洁(如:仅清除残留在设备表面的物料并不进行清洗)。如果生产工艺涉及较剧烈的反应或操作条件,前批残留的少量物料(产品)可能产生降解或蓄积,并可能影响最终成品质量的,则换批次生产时要考虑进行彻底清洁,或者经过验证。
从工艺和设备角度来看,如果企业采用密闭程度较高的生产设备,且工艺中有浓配和稀配,浓配的工艺目的是降低所用原料的微生物、微粒、热原(细菌内毒素)等负荷负,则可将浓配操作放在D级,但稀配操作应至少放在C级。
总之,企业应当根据产品特性、工艺和设备等因素,综合分析,最终确定无菌药品生产用洁净区的级别










 400-6872-158
400-6872-158 点击咨询
点击咨询 





