产品的生产工艺及关键设施、设备应按验证方案进行验证。当影响产品质量的主要因素,如工艺、质量控制方法、主要原辅料、主要生产设备等发生改变时,以及生产一定周期后,应进行再验证。药品生产过程的验证必须包括空气净化系统,制药企业应自行对洁净室环境验证的周期制定一个管理规程。

由ISO14644 中可知,在无菌药品洁净室确认的确认时,主要包括: 已安装的过滤器系统的检漏和完整性测试、气流测试(流速、风量) 及换气次数、自净及恢复时间测试、压差测试、气流方向测试和气流流型研究、温度测定测试、相对湿度测试、悬浮粒子测定、微生物污染监测(浮游菌、沉降菌和表面微生物) 等。洁净室的确认目的是评估洁净室的洁净级别是否满足其预期用途的过程,通常会结合生产需求进行风险评估,测试时需考虑“静态”测试及“动态”测试。在实际测试过程中,设计确认应当证明设计符合用户需求,并有相应的文件。安装和运行确认完成并符合要求后,方可进行性能确认。
根据欧盟附录一4.23用于无菌产品生产的洁净室和洁净空气设备,如单向流单元(UDAFs) 、RABS和隔离器,应根据所要求的环境特性进行确认。每个生产操作要求具有合适的动态下环境洁净水平,以最大程度降低所处理的产品或物料的污染风险。应维持“静态”和“动态”下的适当洁净度水平。可知现阶段对无菌环境的要求更加的贴合实际情况来验证,要求相较更加严格,覆盖更加的广泛,故而在实际操作中的动作影响应多加进行风险评估。

一般说来,空气净化系统在新建、改建之后必须全面验证;正常运行后,应做日常的监测记录工作,如房间的温湿度、风压,以及定期检查微生物和微粒。

空调净化系统中的空气平衡工作是一项技术性较强、调试复杂的工作;一经调整,平时不可随意变动风阀位置,若发现风压流向不对,应找出原因后,才能调整风阀,以免破坏空气的平衡,尤其是无菌生产区域,房间多、洁净级别不同,风压差逐步降低,任何风阀位置的变动,都会引起各房间风压的连锁反应。空调净化系统调试完毕后,应定期检查风量,并计算出各房间的换气次数。风量的检查可每年1~2次。根据积累的验证参数,科学地合理地确定一个环境验证周期。这是确定洁净室环境验证周期的原则。

GMP对无菌药品生产环境要求较严,除空调净化系统安装结束做验证外,每年还要定期测试一些项目:

2)高效过滤器调换或修理后,必须做DOP泄漏试验;
3)空调净化系统的风量每年检查1~2次,并核算出各房间的换气次数;
4)对于洁净级别百级到C级的房间,在无菌产品生产期间,每天应测定悬浮粒子数,不过采样量及采样数目可以按评估减少;
5)浮游菌或沉降菌在无菌产品生产期间,每天应测定,但采样量及采样数目可以按评估减少;
6)表面污染测试在无菌药品生产期间每天应进行;
7)无菌药品在停止生产、空调净化系统关闭后,要恢复生产,需按验证要求重新进行悬浮粒子、浮游菌或沉降菌的测试。

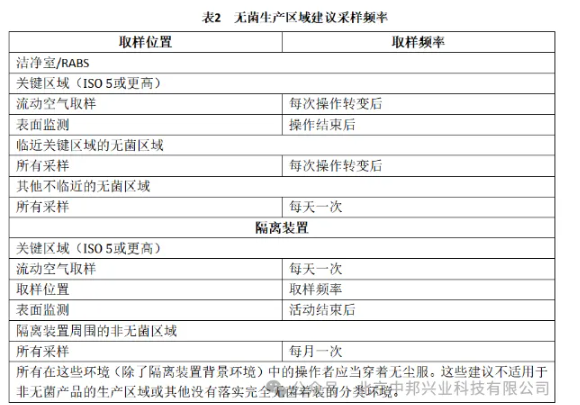
下表给出无菌生产区域日常监测的内容。制药企业可结合企业实际、结合GMP要求实施:
| 无菌生产区域日常监测内容表 | ||||
| 项目 | 洁净度 | 浮游菌 | 表面细菌污染 | 人体细菌污染 |
| 频率 | 百级(每个层流罩下) | 每班1个样品 | 每班3个样品 | 每班从任一操作工身上取样 |
| C级(房间) | 每班1个样品 | 每个房间每个周期3个样品 | 轮流取样 | |
| 位置 | 百级 | 关键操作工艺口处 | 任意取样 | 任意取样 |
| C级 | 工作面处 | 墙、天花板及非接触药粉的设备处任意取样 | 从在该洁净区域工作的操作工中取样 | |
| 采样方法 | —— | 浮游菌采集器 | 培养皿或棉球擦抹法 | 培养皿或棉球擦抹法 |
| —— | 至少25cm2 | 手套2个手指表面及25cm2外表面 | ||
如果您有洁净室洁净度确认方面的问题,或者洁净环境验证类的仪器设备需求,可以直接找北京中邦兴业,专业技术工程师随时可以帮您解答介绍。










 400-6872-158
400-6872-158 点击咨询
点击咨询 





